Tuesday, October 18, 2011
Thursday, September 22, 2011
DIY Bioinformatics: A Whole New Galaxy
This blog post was first published by my company on June 16, 2011
Inspired by Will’s recent book review of Biopunk and the analysis crowd sourcing of the European E. Coli outbreak, I thought I would take another look at DIY (Do It Yourself) biology in this week’s post. Unlike some, I have no interest in trying to run molecular biology experiments out of my kitchen. As anyone who has had the misfortune of trying my cooking would tell you, if there is a way to make a PCR reaction dry and tasteless I'm sure I'd find it. DIY bioinformatics I find more intriguing. I'm not a practitioner as I'm too busy with PSTDIFY (Pay Somebody To Do It For You) bioinformatics, but I like the vision of the lone, amateur scientist, sitting amongst a pile of empty pizza boxes and Red Bull cans finding unknown biological treasure with just their laptop, curiosity and some serendipity. This vision is not so unlikely. Large biological data sets are readily available including thousands of microarray experiments, genotypes and even full genomes. Someone modestly adept at programming or a package like R can interrogate, correlate and mine this data - and indeed this is happening all the time. What about the true amateur, however, who even lacks programming skills? Can the Excel Warrior or my web savy grandma participate in their own DIY bioinformatics adventure? That’s what I set out to discover this week. As a test, I went back to a favorite paper of mine by Majewski and Ott (Genome Research, 2004). What I like about the paper is the number of insights made simply through careful mining of genomic databases. For example, even with inherently noisy data sets like dbSNP and the annotated human genome, the authors were able to clearly see the extent and importance (for splice regulation) of sites near exon-intron boundaries simply by looking at the overall frequency of SNPs discovered in these positions compared to other sites. This figure (F2) from the paper shows the low SNP frequency in the immediate 5' and 3' positions of the intron where it meets the neighboring exons. My test was to see if I could reproduce at least a part of this analysis by simply using free public tools and without programming. I settled on the web-based analysis tool Galaxy as it seemed to have a lot of the functionality I would need and I wasn’t very familiar with it - making me a better stand in for the Red Bull-intoxicated amateur scientist. After some time poking around, I settled on these steps in Galaxy:
My test was to see if I could reproduce at least a part of this analysis by simply using free public tools and without programming. I settled on the web-based analysis tool Galaxy as it seemed to have a lot of the functionality I would need and I wasn’t very familiar with it - making me a better stand in for the Red Bull-intoxicated amateur scientist. After some time poking around, I settled on these steps in Galaxy:
- Get introns from chromosome 12 via UCSC’s Table Browser (I just did chromosome 12 to keep my data sets manageable for this example).
- Get all SNPs from chromosome 12
- Join the introns and SNPs producing a table of only those SNPs that fall within an intron
- Calculate the position of the SNP relative to the 5’ end of the intron
- Count up number of SNPs found at each 5’ position
- Sort results by position (probably not necessary)
- Limit results to just positions within 50 bp of the exon-intron boundary
- Plot the SNP frequency vs SNP location
 This corresponds pretty well to the left portion of Majewski and Ott's own intron plot and was finished before cracking my second can of Red Bull. Score one for DIY!
Before I quit my day (and night) job to make room for the waves of empowered amateurs, it's worth pointing out a few minor details. First, this rudimentary analysis glosses over many important details (such as normalizing for intron lengths) and any publication ready workflow would be much more complex. Second, like all tools of this type, Galaxy walks a fine line to balance functionality and usability. It took me quite a bit of exploring to find the right functions and many of these functions probably only made sense to me because I knew a lot about programming, databases, genomics, etc. Third, it's near impossible to match the power, speed and flexibility a programmer has to analyze data with a web based tool like Galaxy. And finally, although I am empowered by Galaxy to do the steps, the know-how of what questions to ask and the science to understand the observations I make comes from many years of experience - unfortunately there's no short cut around that.
With that said, Galaxy has some very nice features and is a powerful addition to the DIY's tool box. Stock up on your Red Bull now.
This corresponds pretty well to the left portion of Majewski and Ott's own intron plot and was finished before cracking my second can of Red Bull. Score one for DIY!
Before I quit my day (and night) job to make room for the waves of empowered amateurs, it's worth pointing out a few minor details. First, this rudimentary analysis glosses over many important details (such as normalizing for intron lengths) and any publication ready workflow would be much more complex. Second, like all tools of this type, Galaxy walks a fine line to balance functionality and usability. It took me quite a bit of exploring to find the right functions and many of these functions probably only made sense to me because I knew a lot about programming, databases, genomics, etc. Third, it's near impossible to match the power, speed and flexibility a programmer has to analyze data with a web based tool like Galaxy. And finally, although I am empowered by Galaxy to do the steps, the know-how of what questions to ask and the science to understand the observations I make comes from many years of experience - unfortunately there's no short cut around that.
With that said, Galaxy has some very nice features and is a powerful addition to the DIY's tool box. Stock up on your Red Bull now.
Tuesday, September 6, 2011
Excel Hacks: Calculating Oligo Temperature with the Wallace Rule
Calculating oligonucleotide melting temperatures using the Wallace-Itakura formula (2*(number of A's and T's) + 4*(number of G's and C's)) in Excel is a piece of cake. I rarely use it myself as I typically use nearest neighbor with SantaLucia thermodynamic parameters for oligo design, but on occasion it does come up.
Assuming your oligo sequence is in A1, this formula will calculate the Wallace TM:
=2*(LEN(A1)-LEN(SUBSTITUTE(SUBSTITUTE(A1,"A",""),"T","")))+4*(LEN(A1)-LEN(SUBSTITUTE(SUBSTITUTE(A1,"G",""),"C","")))The formula counts the numbers of A's and T's by making a new oligo sequence with A's and T's removed. It then compares the length of the original oligo to the new one - the difference is the number of A's and T's. Counting G's and C's is done in the same manner. Any base other than A,C, G or T will be ignored in the calculation.
Friday, March 4, 2011
Excel Hacks: Reverse Complement a Nucleotide Sequence
Reverse complementing a nucleotide sequence in Excel turns out to be really ugly - in fact, this may be one case where it's better to do some cut and pasting into a web tool or finally go pick up that Perl book that's been sitting on your shelf. However, if you’re desperate and absolutely have to do it in Excel, here's how you can reverse complement short (~30 base) nucleotide sequences.
Complementing
Let's break reverse complementing into its two steps - reversing the DNA sequence and then taking its complement. We'll start with complementing:
As in previous posts, we assume the sequence to be complemented is in cell A1.
This rather lengthy formula is performing a two-step substitution. First, nucleotides are converted to numbers, e.g. “A” to “1”, “C” to “2”, etc. Next, the numbers are then replaced with the complementary nucleotide, so “1” goes to “T”, “2” to “G” and so on. Doing the substitution in two steps allows us to distinguish which bases have been complemented from those that have not. The formula works correctly for nucleotides “A”, “C”, “G”, “T” and “N”. Other degenerate IUPAC codes could be handled, but we’ll leave this as an exercise for the reader.
Reversing
Surprisingly, reversing the DNA sequence turns out to be even trickier. Most programming languages have a function to reverse text, but not Excel. Breaking out a macro in Excel might be worthwhile since writing a little Visual Basic to reverse text isn't too bad. Unfortunately, this is not possible for folks with Macs using Office 2008 since macro functionality was removed.
For a general method to reverse our sequence, our options are limited and ugly. Perhaps one of the less awful methods is repeated use of the MID function. Assuming that our complemented nucleotide sequence is in cell B1, we would do something like this:
The formula constructs the reverse sequence base-by-base up to 30 bases. Make certain that your sequence to reverse is 30 or fewer nucleotides, otherwise your sequence will be truncated without warning. You could extend the function to handle larger sequences, but clearly this method is only practical for short sequences.
Complementing
Let's break reverse complementing into its two steps - reversing the DNA sequence and then taking its complement. We'll start with complementing:
= SUBSTITUTE( SUBSTITUTE( SUBSTITUTE( SUBSTITUTE( SUBSTITUTE( SUBSTITUTE( SUBSTITUTE( SUBSTITUTE( A1, "A", 1), "C", 2), "G", 3),"T", 4), 1, "T"), 2, "G"), 3, "C"), 4, "A")
As in previous posts, we assume the sequence to be complemented is in cell A1.
This rather lengthy formula is performing a two-step substitution. First, nucleotides are converted to numbers, e.g. “A” to “1”, “C” to “2”, etc. Next, the numbers are then replaced with the complementary nucleotide, so “1” goes to “T”, “2” to “G” and so on. Doing the substitution in two steps allows us to distinguish which bases have been complemented from those that have not. The formula works correctly for nucleotides “A”, “C”, “G”, “T” and “N”. Other degenerate IUPAC codes could be handled, but we’ll leave this as an exercise for the reader.
Reversing
Surprisingly, reversing the DNA sequence turns out to be even trickier. Most programming languages have a function to reverse text, but not Excel. Breaking out a macro in Excel might be worthwhile since writing a little Visual Basic to reverse text isn't too bad. Unfortunately, this is not possible for folks with Macs using Office 2008 since macro functionality was removed.
For a general method to reverse our sequence, our options are limited and ugly. Perhaps one of the less awful methods is repeated use of the MID function. Assuming that our complemented nucleotide sequence is in cell B1, we would do something like this:
= MID(B1,30,1) & MID(B1,29,1) & MID(B1,28,1) & MID(B1,27,1) & MID(B1,26,1) & MID(B1,25,1) & MID(B1,24,1) & MID(B1,23,1) & MID(B1,22,1) & MID(B1,21,1) & MID(B1,20,1) & MID(B1,19,1) & MID(B1,18,1) & MID(B1,17,1) & MID(B1,16,1) & MID(B1,15,1) & MID(B1,14,1) & MID(B1,13,1) & MID(B1,12,1) & MID(B1,11,1) & MID(B1,10,1) & MID(B1,9,1) & MID(B1,8,1) & MID(B1,7,1) & MID(B1,6,1) & MID(B1,5,1) & MID(B1,4,1) & MID(B1,3,1) & MID(B1,2,1) & MID(B1,1,1)
The formula constructs the reverse sequence base-by-base up to 30 bases. Make certain that your sequence to reverse is 30 or fewer nucleotides, otherwise your sequence will be truncated without warning. You could extend the function to handle larger sequences, but clearly this method is only practical for short sequences.
Wednesday, February 2, 2011
Populate LinkedIn Publications using PubMed IDs
Note: The LinkedIn-PubMed script has been updated and is discussed here.
LinkedIn now gives you the ability to add publications to your LinkedIn profile. Currently, each publication must be added manually including title, author list, data, abstract, etc. which can be a real drag if you need to enter more than a couple publications. If you're a Firefox user, you can use this simple script to fill in publication data by entering only a PubMed ID.
To install the script:
Here are screen shots of what the LinkedIn page looks like before you install the script:
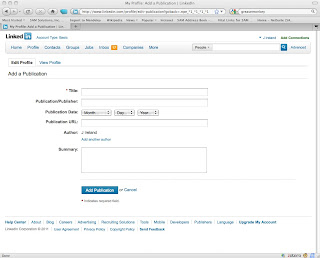
and after:
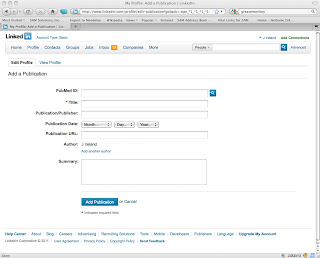
Here's what the form looks like after submitting a PubmedId:
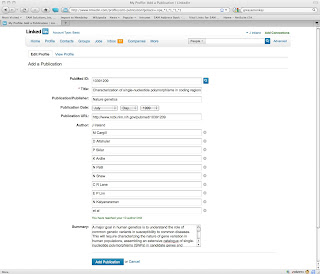
Some Technical Details
I imagine it's only a matter of time before LinkedIn adds PubMed look up or an equivalent convenience, so I didn't want to invest a huge amount of time in this endeavor. GreaseMonkey was a natural choice as it's designed for the task of enhancing existing web sites and doing mash-ups. In this case, I needed to make calls out to Entrez's EFetch, parse the XML response and populate the fields in the LinkedIn form. GreaseMonkey made the PubMed look up a snap, as its GM_xmlhttpRequest method allows for cross-site http requests, something normal JavaScript disallows for security reasons. GreaseMonkey scripts are written in JavaScript and I also brought in jQuery to facilitate manipulating the website's DOM.
As it turned out, the hardest part of the whole project was dealing with the author list. First, LinkedIn automatically includes the LinkedIn user's name in the authors list, so I needed to try to identify and remove the user in the authors reported by PubMed so that it wouldn't appear twice. My simple minded check has been working for my publications, but will surely slip up in some harder cases. The second problem was working with LinkedIn's own JavaScript used to make the author list dynamically increase or decrease as authors get added. In the end, blowing away their scripting and replacing it with my own seemed like the most expedient route. I borrowed heavily from their code to do this and the end result is a bit of an ugly jQuery/straight DOM jumble.
I was able to successfully enter all my publications, but I am certain there will be instances where it falls flat. Let me know how it works for you!
LinkedIn now gives you the ability to add publications to your LinkedIn profile. Currently, each publication must be added manually including title, author list, data, abstract, etc. which can be a real drag if you need to enter more than a couple publications. If you're a Firefox user, you can use this simple script to fill in publication data by entering only a PubMed ID.
To install the script:
- Install the GreaseMonkey Firefox extension by going here and hitting "Add to Firefox".
- Install the LinkedIn-PubMed ID Lookup script by going here and hitting "Install".
- Edit your profile on the LinkedIn site and go down to the link that says "Add a publication". This link will come after your "Experience" section.
- You should see at the top of the form an input box for PubMed ID. Enter your id and hit the search button. The form should be filled out with the publication information as acquired from PubMed. If you're happy with the results, hit submit. Otherwise, edit to your liking and hit submit.
Here are screen shots of what the LinkedIn page looks like before you install the script:

and after:

Here's what the form looks like after submitting a PubmedId:

Some Technical Details
I imagine it's only a matter of time before LinkedIn adds PubMed look up or an equivalent convenience, so I didn't want to invest a huge amount of time in this endeavor. GreaseMonkey was a natural choice as it's designed for the task of enhancing existing web sites and doing mash-ups. In this case, I needed to make calls out to Entrez's EFetch, parse the XML response and populate the fields in the LinkedIn form. GreaseMonkey made the PubMed look up a snap, as its GM_xmlhttpRequest method allows for cross-site http requests, something normal JavaScript disallows for security reasons. GreaseMonkey scripts are written in JavaScript and I also brought in jQuery to facilitate manipulating the website's DOM.
As it turned out, the hardest part of the whole project was dealing with the author list. First, LinkedIn automatically includes the LinkedIn user's name in the authors list, so I needed to try to identify and remove the user in the authors reported by PubMed so that it wouldn't appear twice. My simple minded check has been working for my publications, but will surely slip up in some harder cases. The second problem was working with LinkedIn's own JavaScript used to make the author list dynamically increase or decrease as authors get added. In the end, blowing away their scripting and replacing it with my own seemed like the most expedient route. I borrowed heavily from their code to do this and the end result is a bit of an ugly jQuery/straight DOM jumble.
I was able to successfully enter all my publications, but I am certain there will be instances where it falls flat. Let me know how it works for you!
Subscribe to:
Posts (Atom)